4 Charakteristische Röntgenstrahlung als analytisches Werkzeug [3:20:39]
Mit dem zuvor gewonnen Rüstzeug lernen wir nun, wie wir die Zusammensetzung von Proben bestimmen können. In einem zunächst kurzen Kapitel wird das Energie-Dispersive Spektrometer (EDS) vorgestellt. Anschließend lernen wir die wichtigste und zentrale Grundlage der Winkel-Dispersiven Spektrometerie: die Selektion der Röntgenstrahlung eines Elements mit Hilfe der Bragg-Bedingung, sowie der Rowland-Kreis Geometrie.
Diese Einheit beschäftigt sich mit dem Röntgendetektor. Diese Gaszähler erlauben es Röntgenstrahlung höherer Ordnung auszuschließen, was sehr praktisch ist, um damit Interferenzen zu unterdrücken. Das Thema ist nicht ganz unkomplex, daher ist die Gesamtzeit der Videos zwar etwas kürzer, jedoch müssen besonders die beiden Kapitel/Videos 4.2.4 & 4.2.6 sehr gut studiert werden, um das Thema wirklich zu verstehen. Es wäre nicht erstaunlich, wenn es diesmal im Besondern nötig wäre, zwischendurch anzuhalten, und sich die Dinge durch z.B. eigene Skizzen selbst noch einmal klar zu machen.
Ein zentrales Element um Proben zu messen, ist die Standardisierung, welche wir in dieser Einheit kennen lernen. Wir lernen außerdem erste, konkrete Parameter und Bedingungen kennen, mit denen eine Probe gemessen werden kann, und einige Dinge wie Matrix-Korrektur, Nachweisgrenze, etc. die für eine erfolgreiche Messung beachtet werden müssen.
Im letzten Teil von Kapitel 4 schauen wir uns noch Interferenzen und den Chemical Peak Shift, welche die Messung beeinflussen können. Dann wollen wir jedoch wissen, was und wie wir mit der Mikrosonde oder dem Rasterelektronen-Mikroskop messen können. Schließlich gibt es ein wichtiges Video dazu, was man überlegen sollte oder muss, bevor man am Messtag im Labor steht. Wichtig ist das Video, da es Dir hilft eine erfolgreiche Mess-Kampagne durchführen.
4.1 Energie-Dispersive Analytik (EDS/EDX) [14:15]
Die EDS ist sehr geeignet für eine schnelle, qualitative Analyse, da das gesamte Spektrum abgebildet wird und erste Peaks in Sekunden auftauchen. Die Auflösung ist jedoch vergleichsweise schlecht. Es ist auch eine semi-quantitative Standardisierung möglich, die häufig bei SEMs eingesetzt wird. Eine vollständige Standardisierung ist nur in Ausnahmefällen sinnvoll, z.B. wenn nur ein SEM mit EDS zur Verfügung steht. Es sind weiter z.B. Element-Maps möglich.
- Video
Ja, semi-quantitativ, indem ein Element (z.B. Cu kalibriert wird), es ist auch eine vollständige Standardisierung möglich.
✗Maximal 5
✗Diese werden in einem Periodensystem ausgewählt
✓Alle gleichzeitig
✗Die zehn häufigsten Elemente
✓wahr
✗falsch
✓wahr
✗falsch
4.2 Selektion einer Wellenlänge mit Hilfe der Bragg-Bedingungen (Bragg-Gleichung) [17:13]
Bei WDS-Analysen wollen wir nur die Wellenlänge des Röntgenquants eines Elements messen. Diese können wir mit Hilfe von Analysator-Kristallen und der Bragg-Bedingung selektieren und in den Detektor lenken. Es müssen womöglich Röntgenlinien höherer Ordnung beachtet werden.
Man möchte nur die Röntgenquanten eines Elements zählen, weshalb man die Wellenlänge dieses Elements selektiert, bzw. alle anderen versucht auszublenden.
n * λ = 2d * sin θ – & s. Video
✗1/2 λ
✓1 λ
✓2 λ
✓wahr
✗falsch
✓wahr
✗falsch
4.3 Erfüllung der Bragg-Bedingung mit Hilfe des Rowland-Kreises [14:53]
Die Bragg-Bedingung ist nur in einem Punkt erfüllt. Das würde zu sehr geringen Zählraten führen. Mit gebogenen Kristallen und etwas Geometrie wird es möglich die gesamte Fläche eines Analysator-Kristalls für die Bragg-Beugung, bzw. die Selektion einer gewünschten Röntgenlinie auszunutzen.
In letzter Konsequenz um die gesamte Fläche des Analysator-Kristalls für die Röntgenbeugung auszunutzen.
2:1
✓wahr
✗falsch
✗… hat in der Herstellung Vorteile gegenüber einem flachen Kristall
✓… nutzt den gesamten Kristall für die Röntgenbeugung
✗… fokussiert die Röntgenstrahlen auf die Mitte des Kristall
✗wahr
✓falsch
4.4 Wahl des Analysator-Kristalls [4:09]
Die Bragg-Bedingung wird erfüllt wenn der Einfallswinkel θ sowie der Gitterebenenabstand d stimmen. Tatsächlich reicht nicht ein Kristall mit einem Gitterebenenabstand d aus, um alle gewünschten Wellenlängen zur Beugung zu bringen – oder die Rowlandkreise müssten größer werden, was einen Verlust an Intensität bedeuten würde. Daher werden verschiedene Kristalle mit unterschiedlichen d-Abständen verwendet.
Gitterebenenabstand d und Einfall-/Ausfallswinkel θ – s. Bragg-Gleichung
Kleine Wellenlänge, kleiner d-Abstand.
✓Er hat einen anderen d-Abstand
✗Er hat eine andere Biegung
✗Er hat eine bessere Energie-Aufnahme
✗1
✓2
✗3
✓4
✗pm
✓nm
✗µm
✗mm
4.5 Zählung der Röntgenquanten mit Gas(durchfluss)zählern [7:49]
Nachdem die gewünschte Wellenlänge des zu analysierenden Elements selektiert wurde, können die Röntgenquanten nun gezählt werden. In der Regel kommt dafür ein Gasdurchflusszähler mit einem Gemisch aus 90% Ar und 10% Methan, oder eine geschlossener Xe-Gaszähler zum Einsatz.
ArCH4: offen (Durchflusszähler); Xe: geschlossen
Zur Entladung/Neutralisierung des Ar.
✗1700 eV
✗1700 keV
✓1700 V
✗1700 kV
✓positiv
✗negativ
✓Die Spannung
✗Die Verstärkung
✗Der Strom
✗Der Widerstand
4.6 Peaks, Peaks, Peaks: Satelliten-, höhere Ordnung, Escape-, & Summen-Peaks [18:28]
Der Zähler des Detektors zählt noch insgesamt die Energie von 4 weiteren Prozessen. (i) Die Satelliten-Linien sind den charakteristischen Röntgenstrahlen sehr verwandt, und hängen mit der Probe selbst zusammen. (ii) Peaks höherer Ordnung entstehen, da Röntgenquanten mit der 1/n-Wellenlänge (n: 1, 2, 3 , …) ebenfalls die Bragg-Bedingung erfüllen, wie die gewünschte, zu selektierende Wellenlänge. Diese hängt also mit dem Analysator-Kristall zusammen. (iii) Im Zähler selbst kann Argon angeregt werden, und Röntgenstrahlung aussenden. Entweicht diese aus dem Detektor, fehlt etwas Energie vom zu zählenden Röntgenquant, und es entsteht im Zähler ein zusätzlicher, so genannter Escape-Peak, der diesen geringeren Energie-Impuls abbildet. (iv) Schließlich können zwei gleichzeitig ankommende Peaks als eine Peak mit der dann doppelten Energie gezählt werden. Diese heißen Summen- oder Pile-up Peaks. Die Peaks (iii) und (iv) hängen also mit dem Detektor selbst zusammen.
- oben und Video
Eine Veränderung der Elektronenbindungsenergien/-niveaus durch Leerstellen, Molekül-Orbital-Aufteilungen oder charge-transfer.
✗wahr
✓falsch
✓ja
✗ja: die 1. Ordnung Si mit der 2. Ordnung Nd
✓ja: die 1. Ordnung Si mit der 3. Ordnung Nd
✗ja: die 1. Ordnung Nd mit der 2. Ordnung Si
✗ja: die 1. Ordnung Nd mit der 3. Ordnung Si
✗nein
✗Probe
✗Analysator-Kristall
✓Detektor
✗Zähl-Elektronik
4.7 Zählung der richtigen Impulse (HV, PHA, PHD & SCA) [20:49]
Dem Zählrohr ist eine Zählelektronik nachgeschaltet. Deren wichtigste Funktion ist es, die zusätzlich entstehenden und an sich unerwünschten Röntgenquanten herauszufiltern. Das Konzept hinter dieser Pulshöhen-Analyse (PHA) ist zunächst ein wenig trickreich, jedoch äußerst hilfreich. Die PHA erlaubt zwei Mess-Modi: (i) integral – dabei wird nichts gefiltert, und (ii) differential – nun wird mit Hilfe eines single channel analysers (SCA) gefiltert. Außerdem kann mit der Zählelektronik die Zählrate (Counts per second – cps) optimiert werden,. Dafür wird die Hochspannung (HV) über einen verstellbaren Verstärker (Gain) auf 1700±20 V justiert.
Herausfiltern Peaks höherer Ordnung und/oder Summen-Peaks. (In aller Kürze, ausführlicher wäre besser.)
- oben & Video
✗PHD
✓PHA
✗HV
✗SCA
✗±0,2 V
✗±2 V
✓±20 V
✗±200 V
✗wahr
✓falsch
4.8 Elektronen-Kanone (Electron Gun) & Elektronen-Optik (EOS) [9:17]
Nachdem wir nun fast die gesamte Mikrosonde (und damit auch das SEM) und deren Funktionsweise kennen gelernt haben, schauen wir uns zum Abschluss noch prinzipiell an, wie die Elektronen überhaupt auf die Proben beschleunigt, fokussiert und bei Bedarf darüber gerastert werden.
Sehr knapp: W-Filament und FEG. Unterschiede in Betrieb, Aufbau und schließlich in der Auflösung.
- Video
✗wahr
✓falsch
✗die Filamentspitze
✗die Aperturblende
✓der 1. crossover
✗der 2. crossover
✓5 - 30 kV
✗15 - 30 kV
✗5 - 30 V
✗15 - 30 V
4.9 Messbedingungen: Beschleunigungsspannung (kV), Strahlstrom (nA – µA) & Spot-Size (µm) [6:02]
Nun wollen wir etwas messen. Dazu müssen wir zunächst geeignete Messbedingungen auswählen. Dabei achten wir vor allem auf Beschleunigungsspannung, Strahlstrom und Sp0t-Size, und deren Effekt auf die Probe, bzw. eine Messung.
-> Veränderung der Anregungsbirne
Dass man nicht aus dem umgebenden Harz zu viel Dreck in das Gerät sputtert.
✗15 - 20 µm
✗5 - 15 µm
✓1 - 5 µm
✗1 - 2 µm
✓2 - 4 x 10-8 A
✗20 - 40 µA
✓20 - 40 nA
✓15 und 20 kV
✗15 und 20 keV
✗15 und 20 V
4.10 Standardisierung & k-ratio [7:23]
Um Element-Konzentrationen in einer unbekannten Probe zum messen, vergleichen wir die gemessenen counts per second (cps) eines Elements in der Probe mit den gemessenen cps in einem Standard. Das Prinzip lässt sich sehr leicht über ein Diagramm verstehen. Daraus lässt sich die Standardisierung formalisieren. Die Konzentrationen eines Elements in Probe und Standard sind proportional zu deren gemessenen cps:
\(\frac{c_{i,Probe}}{c_{i,Standard}} \propto \frac{cps_{i,Probe}}{cps_{i,Standard}}\)
c: Element-Konzentration
i: element
cps: counts per second
Die cps werden meist als Intensität ausgedrückt. Das Verhältnis der Intensitäten in Probe und Standard wird als ›k-ratio‹ bezeichnet:
\(\frac{cps_{i,Probe}}{cps_{i,Standard}} = \frac{I_{i,Probe}}{I_{i,Standard}} = k-ratio_{i}\)
Die Proportionalität zwischen Elementkonzentrationen und k-ratio lässt sich über den Korrekturfaktor ZAF nun als Gleichung schreiben:
\(\frac{c_{i,Probe}}{c_{i,Standard}} = k_i \times ZAF_i\)
Die k-ratio wird gemessen, die ZAF-Korrektur lässt sich berechnen, und die Element-Konzentration im Standard ist bekannt, sodass die Konzentration im Element wie folgt berechnet wird:
\(c_{i,Probe} = c_{i,Standard} \times k_i \times ZAF_i\)
- Video
- oben
✗PHA
✓ZAF
✗cps
✗I(std) / I(probe)
✓I(probe) / I(std)
✗cps(std) / cps(probe)
✓cps(probe) / cps(std)
✓Die Steigung der Kalibrationsgeraden im Standard-Diagramm
✓wahr
✗falsch
4.11 Matrix-Korrektur: ZAF, PAP & φ(ρz) [10:17]
Wird ein Element gemessen, interagiert der ankommende Elektronenstrahl, sowie die entstehende Röntgenstrahlung mit den umgebenden Elementen. Dadurch schwächt und verstärkt sich das Signal des zu messenden Elements. Diese Veränderung durch die umgebende, so genannte Matrix muss korrigiert werden. Das geschieht entweder rein theoretisch, indem die drei Einflussparamter (i) Z: mittlere Atomzahl, (ii) A: Absorption und (iii) F: Sekundäre Fluoreszenz berechnet werden (= ZAF-Korrektur). Alternativ wird eine parametrisierte Funktion verwendet (= φ(ρz)-Funktion), welche experimentell bestimmt wurde (z.B. PAP-Korrektur). Tatsächlich messen wir also k-ratios, welche wir dann über eine Matrix-Korrektur in Konzentrationen umrechnen.
- oben & Video
sehr knapp: ZAF – theoretische, φ(ρz) – experimentell parametrisiert
✗Die umgebenden Minerale
✓Die umgebenden Elemente
✗Die umgebenden Komponenten
✗Den Einfluss der Spektrometer-Komponenten
✓Röntgenstrahlung
✗Elektronen
✓Fluoreszenz
✓wahr
✗falsch
4.12 Zählstatistischer Fehler [7:01]
Wenn wir zählen, machen wir meist Fehler. Dieser zählstatistische Fehler wird angegeben mit \(\sqrt{n}\), wobei n die Zahl der gezählten Ereignisse darstellt. Daraus ergibt sich für die Messung von counts ein zählstatistischer Fehler von \(\sqrt{counts}\), bzw. als relativer Fehler \(1/\sqrt{counts}\). Die counts meinen hier alle counts, also cps \(counts imes sec Messzeit\).
- oben & Video
Er wird immer kleiner, irgendwann ist die Abnahme aber nur noch äußerst gering.
✗\(\sqrt{counts}\)
✗\(\sqrt{counts}/1\)
✓\(1/\sqrt{counts}\)
✓\(counts_{ges}=cps imes sec\)
✗\(counts_{ges}=cps / time\)
✓\(counts_{ges}=sec \times cps\)
✗\(counts_{ges}=sec / cps\)
✗1 kcts
✗10 kcts
✓100 kcts
✗1000 kcts
4.13 Untergrund (Background) [3:44]
Bei einer Messung wird nicht nur die charakteristische Röntgenstrahlung, sondern auch ein Teil Bremsberg gemessen. Möglicherweise kommt noch etwas Detektor-Rauschen dazu. Diesen Untergrunde – also primär Bremsberg plus evtl. noch Detektor-Rauschen – wird als Untergrund bezeichnet, und muss vom eigentlich Peak der zu messenden Röntgenlinie abgezogen werden. D.h.: \(cps_{peak} = cps_{gemessen} - cps_{background}\)
v
Bg-Bestimmung links und rechts des Peaks und Korrelation dazwischen.
✗\(counts_{ges}=counts_{peak} - counts_{bg}\)
✓\(counts_{peak}=counts_{ges} - counts_{bg}\)
✗\(cps_{ges}=cps_{peak} - cps_{bg}\)
✓\(cps_{peak}=cps_{ges} - cps_{bg}\)
✗wahr
✓falsch
✗wahr
✓falsch
4.14 Nachweisgrenze (detection limit – d.l.) [8:12]
Der Untergrund wie auch der Peak sind vom Detektor-Rauschen überlagert. Ein Peak kann nur als solcher erkannt und gemessen werden, wenn dieser größer ist als das Detektor-Rauschen. Wir definieren einen Peak als messbar, wenn er größer als das Detektor- und größer als das Peak-Rauschen ist. Daraus ergibt sich für die Nachweisgrenze die prinzipielle Formel:
\(d.l. (Gew\%) = c_{i,Std} \times \frac{3 \times \sqrt{2 \times counts}}{I_{i,Std}} \times ZAF\)
Das Detection-Limit wird direkt auf dem Standard mit gemessen, d.h. \(I_{i,Std}\) wird gemessen, \(c_{i,Std}\) ist bekannt, und \(3 \times \sqrt{2 \times counts}\) sind 3 mal der zählstatistische Fehler des Untergrunds sowie des Peaks. Die statistisch etwas ausgefuchstere Formel für die Nachweisgrenze gibt es im Video.
Dieselbe wie auch zur Bestimmung eines unbekannten Elements. Nur dass statt einer Intensität des unbekannten Elements die Intensität für eine theoretischen Nachweisgrenze eingesetzt wird.
- Video
✗cps
✓Gew%
✓Gew-ppm
✗Gesamt-Counts
✗… der Zählstatistik
✓… des Rauschens des Peaks
✗… der Höhe des Backgrounds
✓… des Rauschens des Backgrounds
✗wahr
✓falsch
4.15 Linienüberlagerungen/Interferenzen [6:46]
Die Energien der Röntgenlinien zweier Elemente können sehr ähnlich sein, da sie entweder nahe beieinander liegen, oder weil die K-Linie des einen Elements eine sehr ähnliche Energie hat wie die L- oder M-Linie eines anderen Elements. Wie auch immer müssen solche Interferenzen genau beachtet werden, um keine falschen Element-Konzentration zu bestimmen.
Wenn sie dieselben/sehr ähnliche Elektronen-Übergangsenergien haben.
- Video
✓Ähnliche Elektronen-Übergangsenergien
✓L- und M-Linien mit ähnlichen keV
✓Eine Überlagerung der Peaks zweier Elemente
✓Unzureichende spektrale Auflösung des Spektrometers
✓… die korrekte Bestimmung der Peak-Lage eines Elements
✓… die korrekte Bestimmung der Element-Konzentration
✓… die korrekte Bestimmung des Untergrunds
✓… die korrekte Bestimmung der cps eine Elements
✗wahr
✓falsch
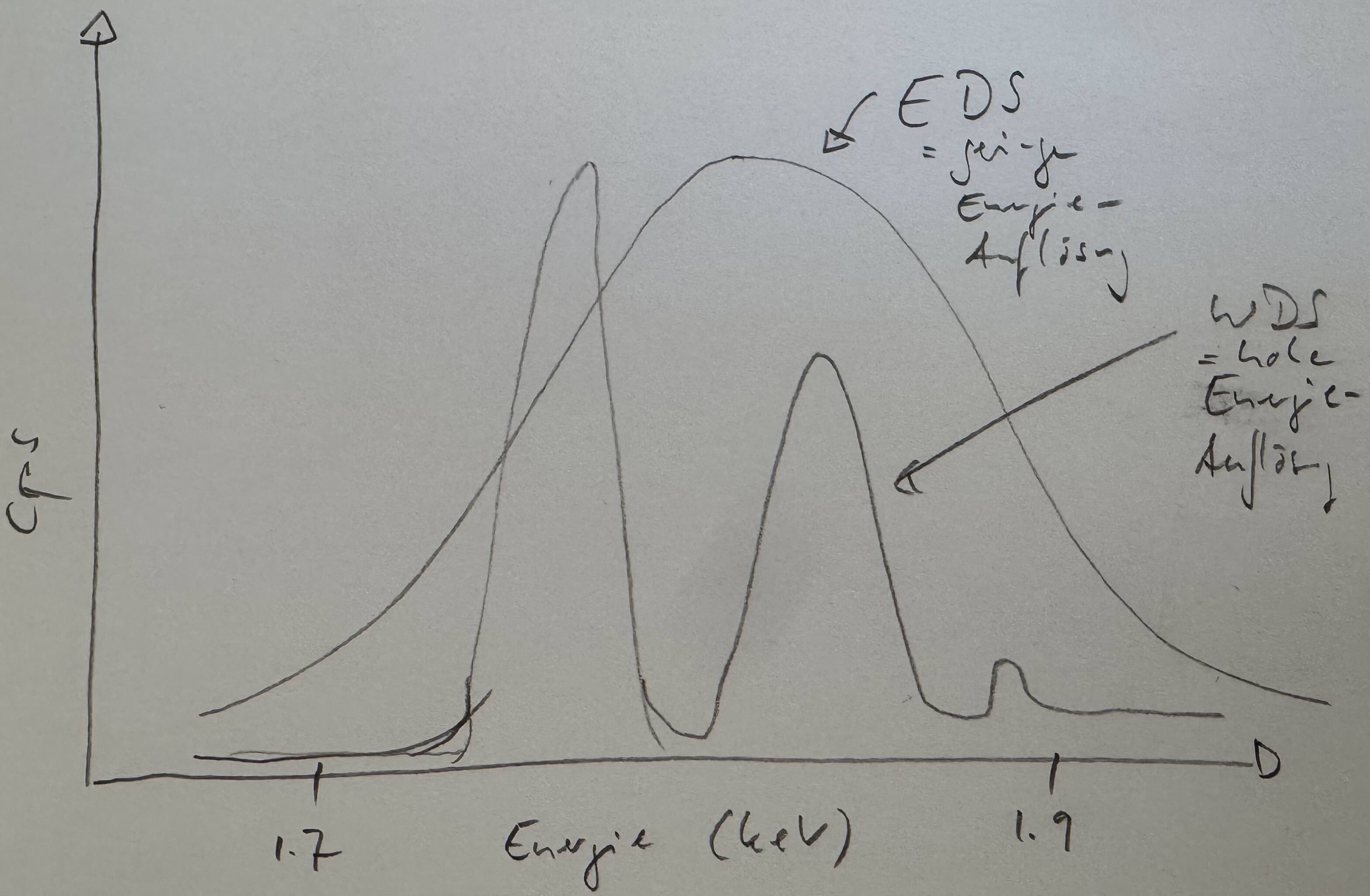
4.16 Energie-Auflösung & Vergleich der Linienbreiten von EDS & WDS [3:59]
Der wohl mit größte Vorteil von WDS gegenüber EDS ist die um Größenordnungen bessere Auflösung von WDS. Während EDS Röntgenlinien im Bereich von 100er eV auflöst, löst WDS im 10er eV Bereich auf. Allerdings hängt die WDS-Auflösung auch vom verwendeten Kristall, sowie dem Durchmesser des Rowlandkreises ab.
Von der Reinheit und Strukturdefekten im Kristall. Ein idealer Kristall könnte die Energie-Auflösung nahe Null bringen, das wäre aber gar nicht wünschenswert, da dann die Einstellung des Peaks schwierig würde.
✗10er keV
✓10er eV
✗100er keV
✗100er eV
✗10er keV
✗10er eV
✓100er eV
✗100er keV
✗EDS
✓WDS
4.17 Chemical Peak Shift [6:52]
Die Elektronen-Bindungsenergien eines Elements können teils sehr unterschiedlich sein, je nachdem in welchem Mineral sie eingebaut sind. D.h., die, Elektronen-Bindungsenergie eines Elements hängt von der Kristallstruktur, sowie den weiteren Elementen im Kristall ab. Die möglicherweise unterschiedlichen Elektronen-Bindungsenergien verschieben den Peak eines Elements mitunter so dramatisch, sodass möglicherweise auf der Flanke des Peaks, statt auf dem Peak selbst gemessen wird. Dann bestimmt man falsche Element-Konzentrationen.
- Video – Diagramm keV vs. Intensität
- Video – Elektronen-Übergangsdiagramm
✗… ändert die cps eines Elements
✓… kann zu Linienüberlagerungen führen
✓… ändert die peak-Lage eines Elements
✗… kann zu einer Erhöhung des Backgrounds führen
✗bis zu 0.1 µm
✓bis zu 0.1 mm
✗bis zu 1 µm
✗bis zu 1 mm
✗wahr
✓falsch
4.18 Punkt-Analysen [8:03]
Mit Punkt-Analysen bestimmen wir die Zusammensetzung unserer Probe. Mit EDS schnell und qualitativ, mit WDS etwas langsamer, dafür quantitativ, bzw. mit hoher Genauigkeit. Es gibt bei Punkt-Analysen ein Reihe von Dingen zu beachten, wie z.B. Nachbar-Minerale, Größe der Probe, Spot-Größe, Genauigkeit der Stage, sekundäre Fluoreszenz oder die EOS-Bedingungen. Als Ergebnis erhalten wir Oxid- oder Element-Konzentrationen. Es ist möglich Punkte zu programmieren und z.B. über Nacht laufen zu lassen.
zB: kV, nA, Spot-Size
Nicht zu klein, da sonst ein Mess-Punkt evtl. nicht sauber angefahren wird.
✗wahr
✓falsch
✗5 min
✗bis zu 15 min
✓2-3 min
✗dauert nur Sekunden
✗100-200 nm
✓1-2 µm
✗4-5 µm
✗1-2 mm
4.19 Linien-Analysen [4:41]
Mit Linien-Analysen bestimmen wir Konzentrations-Profile über Minerale, Gläser – oder ganz allgemein Phasen. Hier müssen wir ebenfalls eine Reihe von Dingen beachten, zunächst oftmals dieselben wie bei Punkt-Analysen, hinzu müssen wir überlegen welche Schrittweite wir brauchen und wie lange so eine Line dann wohl braucht. Lines können natürlich ebenso wie Punkte vorprogrammiert werden.
- Video
✗wahr
✓falsch
✗5 min
✗bis zu 15 min
✗2-3 min
✓dauert nur Sekunden
✗… etwa gleich groß sein wie ein einzelne Punkt-Messung
✓… mind. doppelt so groß sein wie ein einzelne Punkt-Messung
✗… etwa 4x so groß sein wie ein einzelne Punkt-Messung
✓… kommt auf die Anzahl der Messpunkte entlang der Linie an
4.20 Element-Maps [3:09]
Element-Maps sind bei EDS wie auch bei WDS in der Regel qualitativ. Nur sehr selten werden quantitative Grids erstellt. Bei EDS wird für eine Map der Strahl, bei WDS für eine höher Genauigkeit die Probebühne bewegt. Element-Maps können sehr aussagekräftige Übersichten der Elementverteilung in einer Probe geben. Element-Maps brauchen jedoch schnell viele Stunden und sind daher teuer. Werden sei benötigt, lässt man sie gerne z.B. vorprogrammiert über ein Wochenende laufen.
Jeder Wert repräsentiert die Häufigkeit eines Elements an einer gemessenen x-/y-Koordiante.
✗wahr
✓falsch
✓Mineralphasen darzustellen
✓Zonierungen zu erkennen
✗eine schnelle Phasenanalyse zu machen
✓kleine Phasen zu entdecken
✓… der Elektronen-Strahl über die Probe rastert
✗… die Probe sich unter einem ortsfesten Elektronen-Strahl bewegt
4.21 Proben- und Mess-Vorbereitung [18:24]
Wenn man an ein Gerät geht ist es sehr hilfreich im Vorfeld gut zu überlegen was man messen will, bzw. welche Ergebnisse man möchte, und – die Probe entsprechend vorzubereiten. Diese Sachen können oder sollten am Besten auch mit mir im Vorfeld einmal durchgesprochen werden, bzw. gibt es bei mir eine Checkliste, worauf man achten sollte – hier einmal die Checkliste erklärt in einem Video.
Mounts, Dünnschliffe
- oben & Video
✓wahr
✗falsch
✗1
✗3
✓5
✓wahr
✗falsch